Machine Learning and Single-Cell Genomics Online Symposium
The Machine Learning & Single-Cell Genomics online symposium, hosted by Immunai & Fabian Theis from Helmholtz Munich, brought together world leaders to discuss recent breakthroughs that may change our approach to drug discovery and biology in general.
Single-cell (sc) genomics, the study of genetic information in individual cells, examines biology at unprecedented depth, but the growing scale and complexity of sc datasets are bound to exceed human interpretation. On the other hand, the depth of sc profiling coupled with the power of machine learning (ML) has the potential to transform the healthcare system – from diagnosis to patient stratification to drug development.
But every revolution starts small. So did sc genomics with the RNA measurements of just a few dozen cells. Since then, the field has grown exponentially and even recently exceeded the milestone of a million profiled cells. The explosion in dataset size was mostly driven by technological advancements, especially through the commercialization of droplet-based technologies that both democratized sc experiments and established 10x genomics as the leading provider of sc genomics applications.
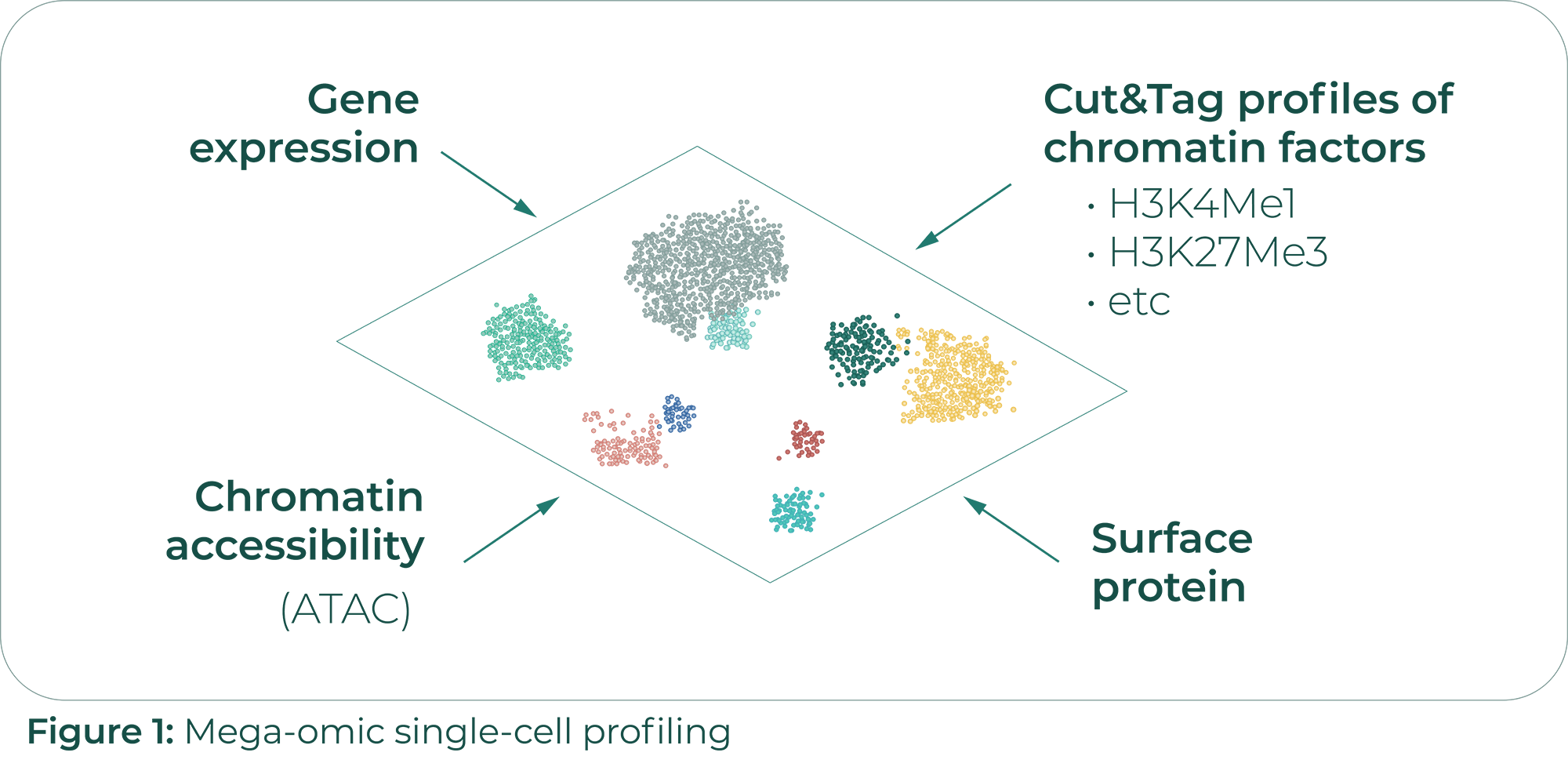
Michael Schnall-Levin, founding scientist of 10x genomics, presented the company’s latest developments. Their next-generation chromium platform (Chromium X) plummets per-cell costs by 70% and increases throughput up to a million cells per single run. Visium HD, a complementary product to link gene expression profiles with a cell’s spatial position, will allow transcriptional mapping of fresh or FFPE tissue sections at 5-micron resolution – less than the average mammalian cell. Despite such remarkable advances, RNA expression is only one piece of the puzzle. Proteins are the cell’s functional unit. Regulatory and lineage commitment decisions often happen at the level of DNA accessibility. Furthermore, the ontogeny and antigen specificity of lymphocytes is encoded in the sequence of their antigen binding receptors. Sc transcriptomics therefore only serves as a proxy for a cell’s biological state. To overcome this limitation, 10x genomics expanded their portfolio and now supports CITE-seq, ATAC-seq, VDJ-seq and even sequencing approaches that identify paired TCR/BCR-antigen complexes at sc resolution (Fig. 1).
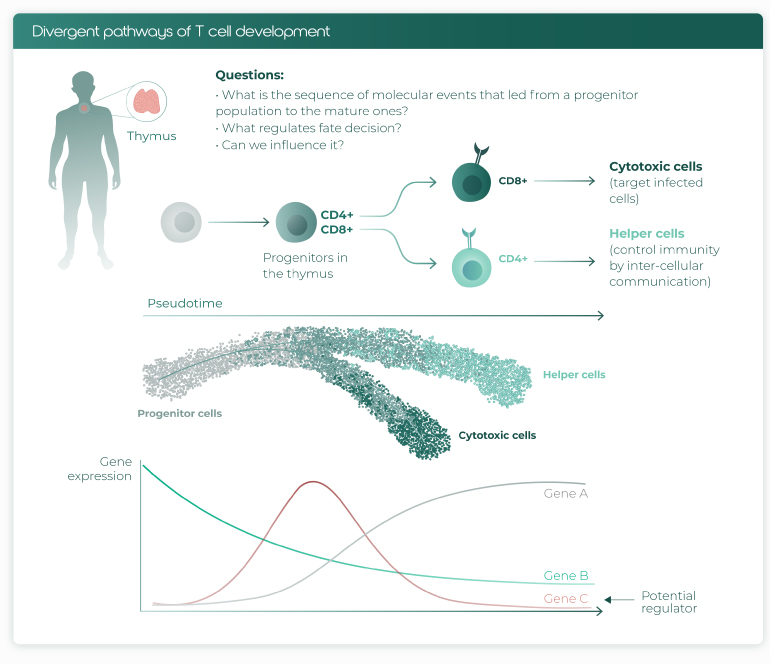
It is technological advancements like these that continue to push boundaries, but such multi-modal DNA, RNA, and protein measurements require dedicated analytical solutions to integrate the different modalities into a joint representation (further discussed here) (scVI, TCR-gex). Nir Yosef, associate professor at Berkeley, pioneered the work on multi-modal data integration which culminated in the software suite scVI/totalVI. The tool already demonstrated its power for the study of T-cell lineage commitment, a process marked by a dense sequence of molecular events. After sampling RNA and protein profiles from differentiating T-cells, totalVI harnessed both data types to order the cells along a differentiation trajectory. This empowered Yosef and his team to identify the regulators of T-cell bifurcation, i.e. the molecular decision-makers that commit T-cells to either the CD4 or CD8 lineage (Fig. 2). Sc multi-omics coupled with proper data integration thereby shed new light on a differentiation process that has fascinated and puzzled scientists for decades.
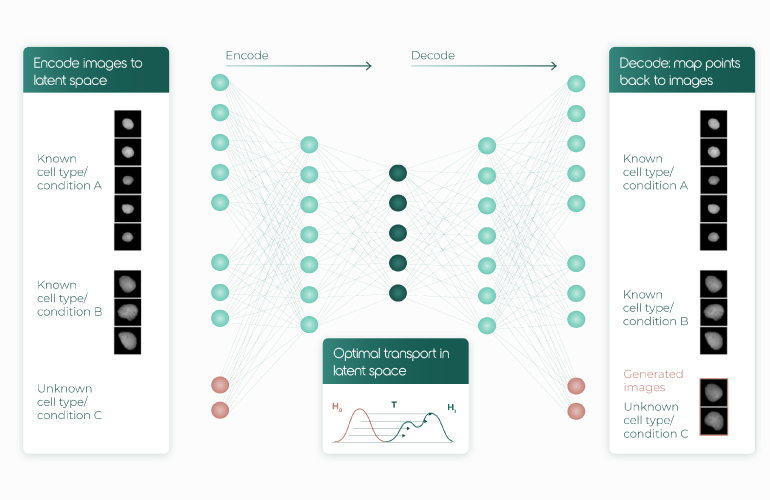
Clearly, sampling the entire spectrum of cellular states is ideal to dissect subtle biology, but such high-density measurements are impractical in the clinic. For example, cancer progression is a continuous process we mostly observe when the tumor has already manifested. To still gain insight into cancer onset and the early molecular events, Caroline Uhler followed a cell line’s malignant transformation (from normal to metastatic) with imaging and sc transcriptome snapshot measurements. She and her team then used autoencoders, unsupervised learning algorithms that find low dimensional embeddings of the data. Under the constraint of minimized transportation cost, Uhler connected the resulting cell embeddings between different stages of malignancy and inferred the most ‘energy-efficient’ path the cells may have taken. We thus get morphological and expression predictions of such transient states without ever measuring them directly (Fig.3)
Uhler didn’t stop there. The next big challenge in biology was already on the horizon: can we train models to predict a cell’s response to perturbations that are untested? Based on her work on ML for SARS-Cov-2 drug repurposing, it doesn’t seem impossible.
Luis Voloch, Immunai’s CTO and co-founder, shared how Immunai leverages all the latest advancements in sc multi-omics, computational power, and ML to change the drug discovery process (including perturbation predictions). He highlighted the inherent match between the layered organization of artificial neural networks and the hierarchy of the immune system (Fig. 4).
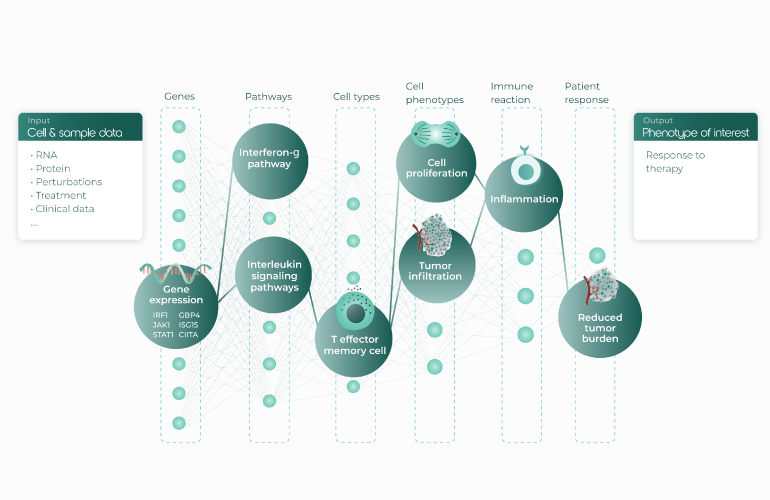
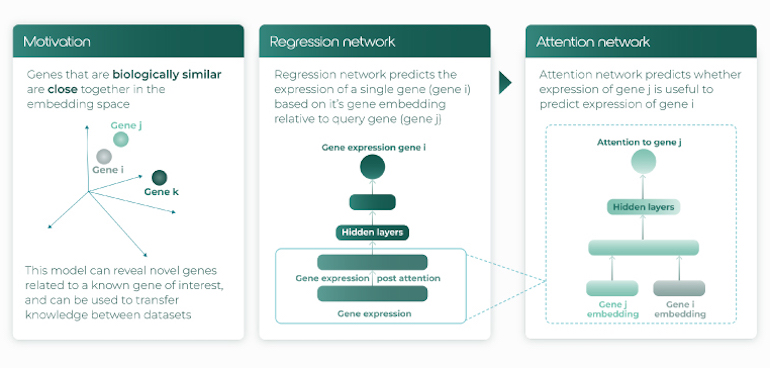
Immunai applies multitask and transfer learning to their immunomic database AMICA, which stores transcriptome, surface protein, and chromatin accessibility data – a detailed library of the immune system’s secret microcosm. With its millions of annotated cells and standardized in vitro and clinical metadata, AMICA-trained annotation algorithms already outperform current state-of-the-art methods for immune cell annotation. Immunai also uses attention networks (Fig. 5) for a more nuanced understanding of gene regulation. Given the unique ability of such networks to uncover how the activity of one gene affects that of others, the approach may become the new gold standard in the field of sc genomics. Putting all these promises to the test, the company’s ML platform has already suggested gene candidates whose modulation improves T-cell function; Immunai is now pursuing several of these candidates as drugs.
The Machine Learning & Single-Cell Genomics online symposium highlighted the field’s drive for innovation and entrepreneurship. The coming years will benchmark our ability to translate scientific breakthroughs into clinical practice, but already now it is clear that the marriage of sc genomics and ML starts to shape a new reality – a reality that had long seemed like science-fiction.
Related posts
Advancements in Gene Regulatory Networks and Perturbation Predictions: Immunai’s Third Symposium