Advancements in Gene Regulatory Networks and Perturbation Predictions: Immunai’s Third Symposium

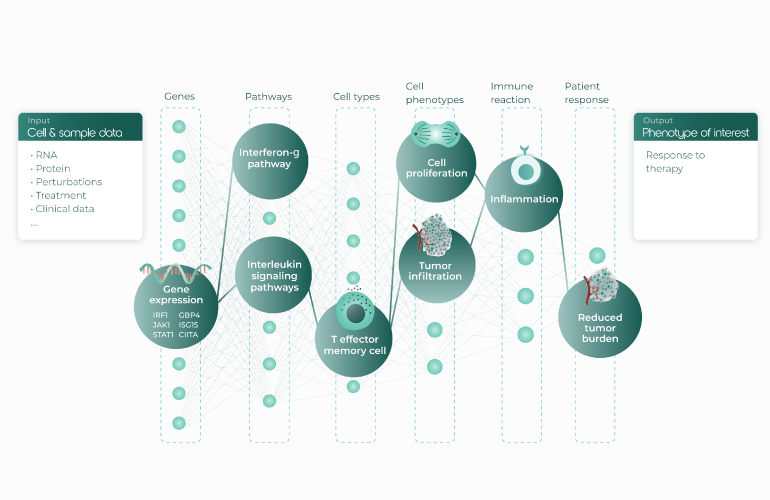
A gene regulatory network (GRN) is a group of interacting molecular regulators within a cell that govern gene expression. Gene expression determines cell function within a biological system, and understanding the regulation of that expression is key to answering fundamental questions in biology and to developing new therapeutics. With recent advances in single-cell biology, we are poised to dramatically improve our understanding of GRNs and translate this knowledge into better treatments for patients.
For Immunai’s third symposium, it brought several scientific luminaries who shared their recent research work related to GRNs. The symposium was moderated by Caroline Uhler, an Associate Professor at Massachusetts Institute of Technology (MIT).
Fabian Theis’s talk opened the symposium [watch Fabian’s talk here]. Fabian is the Head of the Helmholtz Munich Computational Health Center and pioneered many ML methods for single cell biology, such as models for single cell states, trajectories and perturbational responses. Fabian uses a combination of singe-cell sequencing and machine learning to gain a deep understanding of cells, including how they develop, interact, and sometimes contribute to disease.
Fabian presented tools and frameworks that he and his collaborators have developed to advance single-cell research. He began by sharing CellRank, a toolkit to understand cell dynamics using single-cell data. The tool is inspired by and takes its name from Google’s famous PageRank algorithm, as it uses Markov chains to understand relationships between individual cells, similar to PageRank’s use of Markov chains to understand relationships between web sites. CellRank can help users compute initial, terminal, and intermediate macrostates of a biological system, infer cell fate probabilities, visualize gene expression trends along specific lineages, and identify potential driver genes for cellular trajectories.
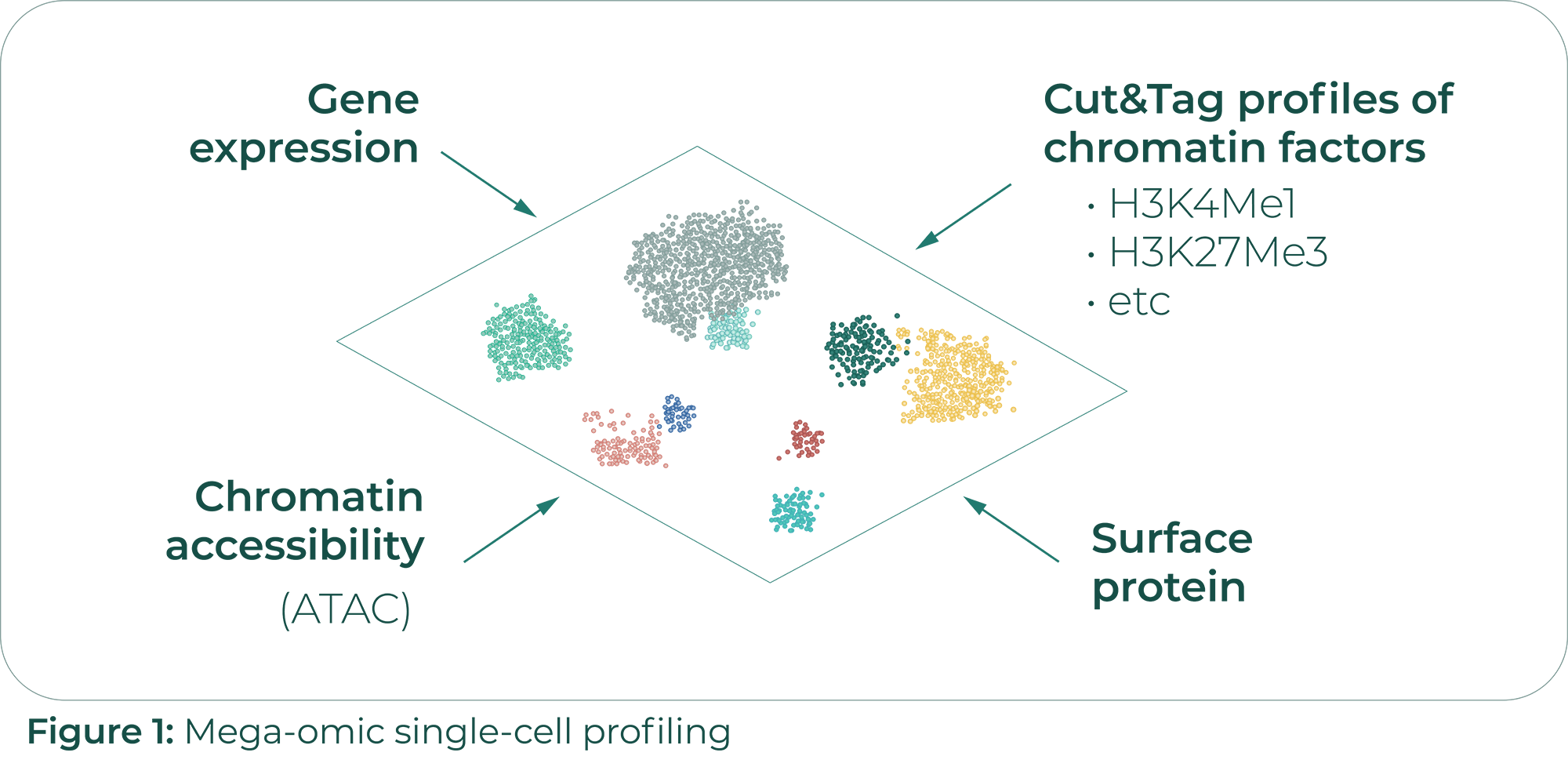
Tools like CellRank bring more value when applied to large-scale data. The richness and variety of data available today is impressive and will continue to increase. We can measure a variety of -omic data including DNA, RNA, protein and metabiomic related data. We can do so at a variety of scales: for organisms, organs, tissues, and single cells. On top of that, there are a variety of perturbations. Altogether, these data can give us many signals that help us understand cell fate in healthy and diseased states.
With respect to learning Gene Regulatory networks from this data, Fabian pointed out that the field has long been using deep learning to learn unsupervised representations of cells with feedforward autoencoders that take gene expression in single cells as input. In a sense, the hidden layers of these neural networks must be learning relationships between genes as they develop an understanding of cells; however, there is much more that deep learning allows us to do in terms of adding constraints to our genomics models. For example, computer vision models using convolutional neural networks are shift invariant. We can also reuse knowledge learned on one dataset in a new context (“transfer learning”). In this vein, Fabian presented two vignettes taking inspiration from deep learning models that perform transfer learning and incorporate constraints.
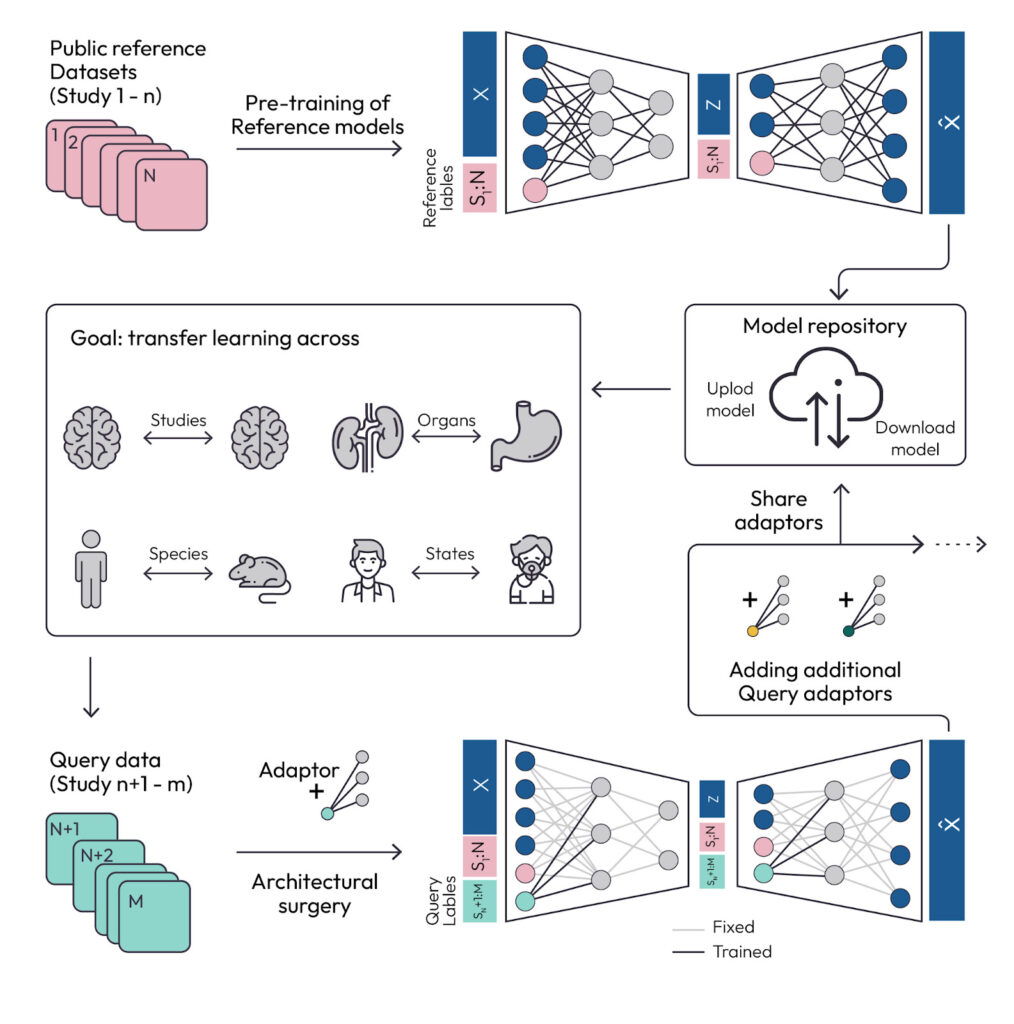
The first example was scArches, a package that helps integrate new single-cell data into reference atlases, exemplifying transfer learning. The tool facilitates collaboration across many groups and teams and enables integration of datasets from different projects. The scArches model makes it easy to build single or multi-modal reference atlases and share the trained machine learning (ML) models and the datasets. Users can download a pre-trained reference model and continue to learn representations of new datasets, or map query datasets onto the reference atlas with the pretrained model (Figure 1). For example, scArches was used to map COVID-19 diseased cells onto a healthy reference atlas, enabling discovery of disease-specific cell states.
The second vignette introduced Explainable Programmable Mapper (expiMap), which uses the aforementioned trick of adding constraints to the model to improve learning efficiency. Instead of the traditional autoencoder, the model incorporates gene programs in the latent space and constrains the model to utilize only some of the gene programs. Surprisingly, the incorporation of this constraint did not diminish the predictive power of the model, while providing an interpretable explanation for which gene programs are running in various cell states. For future research, Fabian believes that this deep learning technique will enable better modeling of perturbational data among other applications.
The second talk was presented by Samantha Morris, an Associate Professor of Developmental Biology and Genetics at Washington University in St. Louis [talk found here]. Her lab is focused on the question of how to morph one cell identity into another, which requires her to understand how cell identity is defined. Because Gene Regulatory Networks are the master regulators of cell identity, they are important for her to understand in attempting to reprogram cells. Samantha and her team developed a platform called CellOracle which dissects cell identity via gene regulatory network inference and in silico perturbation.
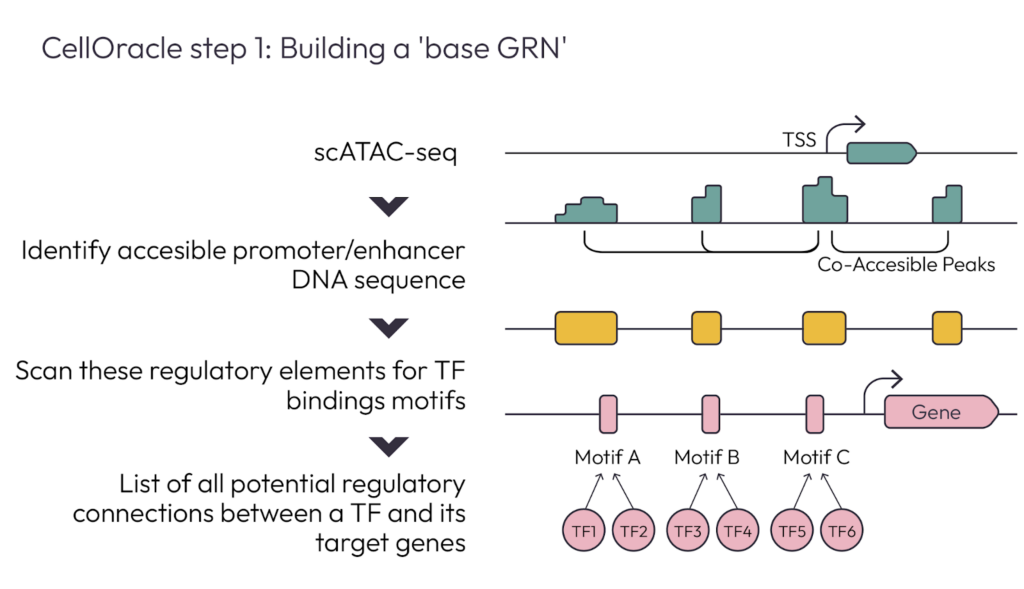
CellOracle is a machine learning based tool that combines single-cell transcriptomic (scRNA-seq) and epigenomic (scATAC-seq) data to infer GRNs. It can investigate how GRNs change during cell reprogramming, differentiation, and development, as well as understand how transcription factors regulate cell identity. CellOracle begins by leveraging single-cell ATAC-seq data to create a Base Gene regulatory network, then uses a regularized machine learning model to refine this GRN by training on scRNA-seq data. Upon completion of the refined GRN, CellOracle can use the network to simulate transcription factor (TF) perturbations.
Samantha and her team wanted to test CellOracle in their research. One experiment they undertook was a single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. They conducted knockout simulations for all TFs that had inferred connections to at least one other gene across zebrafish development. That experiment allowed them to discover new phenotypes and new regulators of axial mesoderm development.
Other research groups are already starting to make use of CellOracle in their own work. For example, Vittoria Bocchi, from the University of Milan and her collaborators are applying CellOracle to ongoing work on building a fetal striatum single-cell atlas to predict the role of Znf467 in medium spiny neuron maturation.
In conclusion, CellOracle infers GRNs and simulates cell identity changes in response to TF perturbations in silico. CellOracle was validated in hematopoiesis and zebrafish development. It can identify new regulators of cell identity as well as new transition factors for precision reprogramming strategies.
The third talk was presented byJason Buenrostro, Assistant Professor, Harvard University (Stem Cell and Regenerative Biology)[talk found here]. Jason led the development of the Assay for Transposase Accessible Chromatin (ATAC-seq), which measures chromatin accessibility in cells. Later, Jason demonstrated single-cell ATAC-seq to map the accessible genome of individual cells. These methods are important tools for the field of epigenomics, which studies the biological mechanisms behind gene regulation and expression. Importantly, single-cell epigenomics can provide insight into cellular potential and memory, as well as early markers of cell type and cell state transition. Pairing single-cell ATAC-seq with RNA data can also enable mechanistic models of gene regulation across heterogeneous cell types, as was demonstrated in Samantha Morris’s talk.
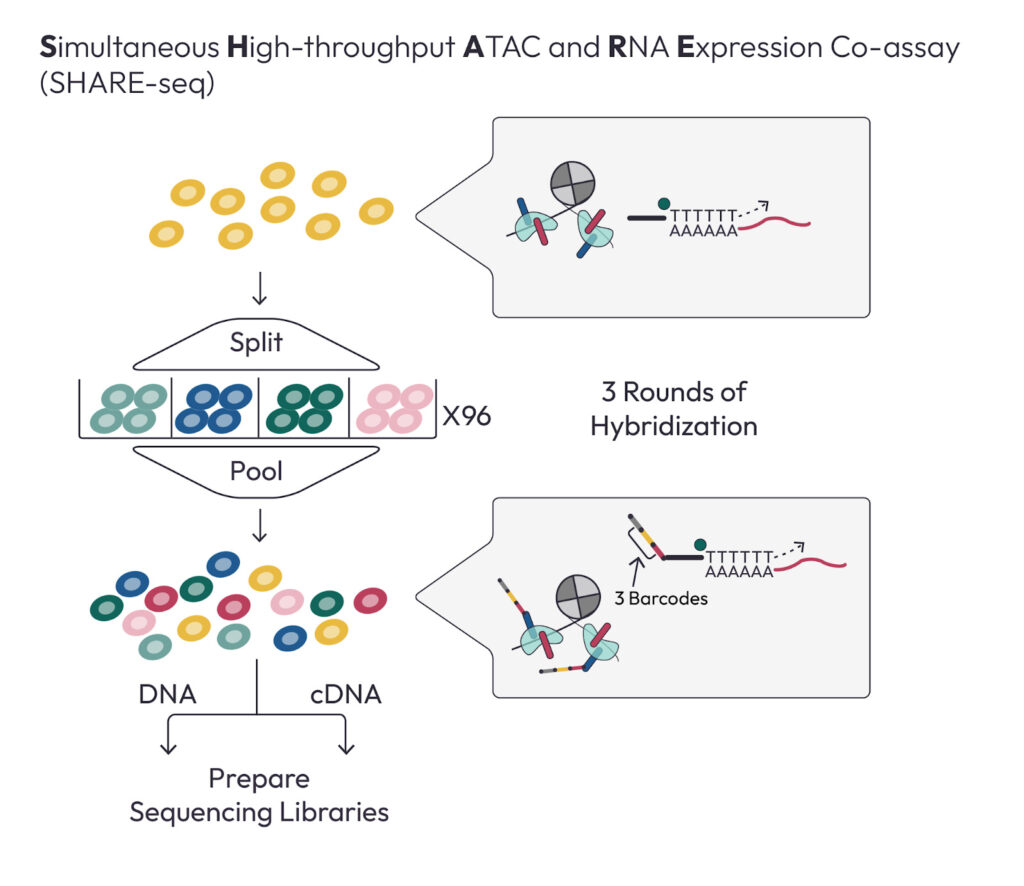
Jason presented a method for Simultaneous High-throughput ATAC and RNA Expression with sequencing (SHARE-seq), led by Sai Ma. SHARE-seq is a highly scalable (to more than 100k cells) approach for measurement of chromatin accessibility and gene expression in the same single cell. Interestingly, SHARE-seq is relatively cheap, with the cost of library preparation varying from $0.001 to $0.04 per cell.
Using SHARE-seq, Jason examined the question of whether chromatin and RNA defined cell types are always the same, and found that they generally are, although there are notable exceptions. Using SHARE-seq to map ATAC-seq peaks to genes also allowed Jason’s group to ask a very simple question: how many ATAC-seq peaks are correlated with each gene? They define genes with many peaks as Domains of Regulatory Chromatin (DORCS), and find that DORCs are often known lineage determining genes. By looking at the dynamics of DORCs, they find that enhancers turn on before promoters, and promoters turn on before gene expression, providing a general model for gene activation in development.
Jason also presented a method for using the shared ATAC and RNA data to empower mechanistic models of gene regulation across different cell types. Motivated by the question of whether ATAC-seq can elucidate the functional substructure of enhancers, Jason built a dataset of approximately 1M human bone marrow cells to focus on hematopoiesis via SHARE-seq. In the ATAC-seq data, he can see the footprint of the nucleosome, and even see it slide around during development exposing DNA for TF binding. This deep mechanistic visibility can help us to understand how multiple regulatory factors work together or even compete for access to DNA.
All three talks demonstrated that improved experimental and computational methods are providing unprecedented insight into the regulatory mechanisms behind gene expression. Machine learning can help us to understand the enormous amount of new data being generated and provides a way to integrate knowledge across studies and experiments. Ultimately, we believe that the insights shared in these presentations will lead to improved biological understanding and better therapies for patients.
Related posts
Immunology and Machine Learning Online Symposium